Celkem je 45 různých nemoci lysozomálního skladování, které jsou heterogenní skupinou vrozených metabolických chorob. Lidé, kteří trpí některým z těchto onemocnění, mají genetickou vadu. Všechna onemocnění při skladování mají jednu společnou věc: určitý enzym buď chybí, nebo je pouze částečně funkční.
Co je to lysozomální choroba?

© návrhya - stock.adobe.com
Tyto vrozené choroby skladování jsou vzácné, protože je postiženo méně než pět z 10 000 lidí. Různá onemocnění mají velmi odlišný průběh a příznaky se mohou velmi lišit.
Nejslavnější formy onemocnění lysozomálního skladování jsou Fabryho choroba, Gaucherova choroba, Pompeho choroba a mukopolysacharidóza (MPS). Často se označují jako „sirotci medicíny“, protože cesta ke specifické diagnóze a vhodné terapii může být velmi dlouhá. Někdy to může trvat roky, než se postižené dozvědí, co se s nimi děje.
příčiny
Nemoci lysozomálního skladování jsou charakterizovány určitými formami dědičných metabolických chorob. Pacientovi chybí důležitý enzym, který zajišťuje hladký průběh metabolické rovnováhy. V méně výrazné formě není tento enzym přítomen alespoň v dostatečném množství.
Úkolem enzymů je zneškodňovat znečišťující látky a odpadní látky, které se hromadí v lidském organismu prostřednictvím metabolismu přes lysozomy, nebo je znovu zpracovat tak, aby se neobjevily příznaky.
Pokud existuje nedostatek enzymů, tento hladce fungující likvidační cyklus již není zaručen. Škodlivé látky se usazují v buňkách a narušují metabolický cyklus. V počáteční fázi nemají poruchy znatelný účinek, existuje jen několik omezení. Pokud však tato metabolická porucha zůstává neléčena v důsledku nedostatku enzymů, příznaky se znásobí, protože buňky se velmi zvětší.
Příznaky, onemocnění a příznaky
V nejhorším případě se to pod. Důsledky jsou poškození kostí, nervového systému, sleziny, ledvin, svalů nebo srdce. Fabryho choroba způsobuje, že tuk (globotriaosylceramid, Gb3) je v buňkách uložen v důsledku snížené nebo chybějící enzymatické aktivity. Tyto nežádoucí usazeniny mohou vést k silné bolesti v nohou nebo prstech, cévní mozkové příhodě a poškození ledvin.
Diagnóza a průběh nemoci
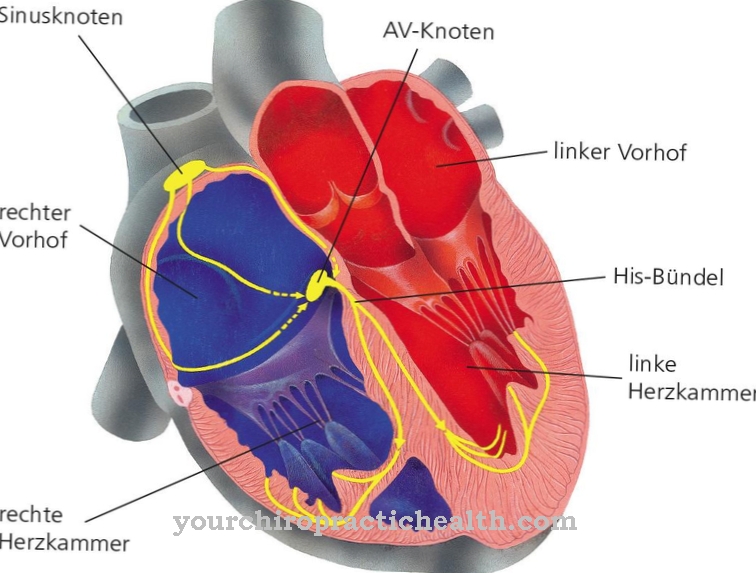
Tento klinický obraz ovlivňuje současně různé systémy: krevní cévy, ledviny, srdce a nervový systém. Autozomálně recesivní dědičná Gaucherova choroba způsobuje mutaci enzymu "beta-glukocerebrosidáza" a vede k akumulaci substrátu v buňkách, zejména v makrofázích (vychytávací buňky), které patří do retikuloendoteliálního systému. Změny krevního obrazu, zvětšení jater a sleziny a zranění kostí.
Nemoc je progresivní a je většinou etnická, protože ve většině případů se vyskytuje u lidí židovského původu. Pompeho choroba je také známá jako „nedostatek kyselé maltázy“. Klinický obraz patří do skupiny glykogeneze typu II. U postižených chybí enzym alfa-1,4-glukosidáza (kyselá maltáza) nebo není k dispozici v dostatečném množství. Pacienti trpí narušením odbourávání glykogenu ve svalech ničením svalových buněk ve formě ukládání cukru.
Mukopolysacharidóza typu I (MPS), známá také jako Hunterova choroba, má různé klinické příčiny. Hurlerova choroba je nejzávažnější formou a Scheieho choroba je na konci klinické patogeneze. Mezi těmito dvěma formami progrese existují přechody různých charakteristik. Nejvýznamnějším rysem je narušené odbourávání uhlohydrátů, které se hromadí v lysozomech buněk.
U pacientů s Hunterovou chorobou se může vyskytnout krátká postava, zvětšená slezina a játra, hrubé rysy, zesílená kůže, zvětšený jazyk a potíže s dýcháním. Kromě toho se kostra často mění v oblasti pánve, páteře, kostí ruky a lebky. Jsou možné pupeční a [[tříselné kýly].
Komplikace
Ve většině případů se příznaky nebo komplikace objevují při této nemoci velmi pozdě. Z tohoto důvodu je diagnostikována pozdě, takže včasná léčba není ve většině případů možná. Bez léčby dochází v průběhu onemocnění k různým stížnostem a poškození vnitřních orgánů.
Obzvláště jsou postiženy ledviny, játra a slezina. Toto onemocnění může být ovlivněno také srdce, které může v nejhorším případě vést k srdeční smrti. Dále dochází k poškození ledvin a postižené osoby často trpí bolestí v prstech na nohou nebo prstech. Paralýza může také nastat, pokud byl tímto onemocněním mozek poškozen. Játra a slezina mohou být zvětšeny a způsobit také silnou bolest.
Není neobvyklé, že kosti postiženého jsou křehké a bolestivé. Léčba tohoto onemocnění se ukazuje jako obtížná. V mnoha případech je délka života postižené osoby výrazně snížena. Při užívání léků obvykle neexistují žádné zvláštní komplikace. Pozitivní průběh nemoci však nelze v každém případě zaručit.
Zde najdete své léky
➔ Léky proti bolestiKdy byste měli jít k lékaři?
Ztráta vlasů, problémy s klouby a poruchy orgánů jsou možné příznaky lysozomálního skladování. Pokud se příznaky stále opakují nebo se objevují náhle, aniž by byla nalezena příčina, doporučuje se navštívit lékaře. Pokud příznaky souvisejí s již diagnostikovanou vadou enzymu nebo jiným závažným onemocněním, měl by být konzultován odpovědný lékař. Neošetřené onemocnění při skladování může vést k demenci, neplodnosti, neuropatií a dalším komplikacím, z nichž některé jsou život ohrožující. Proto by měly být vyšetřeny všechny možné příznaky, i když neexistuje žádné zvláštní podezření.
Příznaky lysozomálního skladování se mohou objevit ve fázích nebo se vyvíjet pomalu, ale vždy vyžadují vyšetření a léčbu. Postižení lidé nejlépe mluví přímo se svým rodinným lékařem nebo internistou. Skutečná terapie se obvykle provádí na specializované klinice pro interní nemoci, přičemž fyzioterapie nebo psychoterapie mohou být spojeny v závislosti na symptomech. Zejména jsou indikována terapeutická opatření kvůli často negativnímu průběhu onemocnění.
Terapie a léčba
V závislosti na tom, jak brzy je provedena adekvátní diagnóza, mohou být tato dědičná onemocnění léčena enzymatickou substituční terapií velmi dobře, takže postižení lidé mají mnohem méně symptomů, a tím i lepší kvalitu života. Tato náhradní terapie se používá podle klinického obrazu.
Lidé trpící Gaucherovou chorobou postrádají „enzym ß-glukocerebrosidázu“, která je biotechnologicky produkována a je infundována do těla pacienta. Lysozomy působí efektivně a jsou schopny absorbovat látky ze svého bezprostředního okolí. Z tohoto důvodu jsou uměle používané enzymy modifikovány tak, že mohou být ideálně dodávány do lysozomů.
Makrofágy (fagocyty) štěpí glukocerebrosidy akumulované v buňkách. Tuto terapii lze srovnávat s inzulínovou terapií u diabetes mellitus s tím rozdílem, že nejde o chybějící hormon, ale o enzym, který není dodáván. Tělo pravidelně štěpí všechny látky, včetně dodaného umělého enzymu.
Vzhledem k tomuto pravidelnému rozkladu látky musí pacienti podstoupit tuto infuzní terapii pravidelně až do konce svého života. Léčba substitučními enzymy nepůsobí symptomaticky, ale přímo bojuje s příčinou dědičného onemocnění. Lékaři nazývají tuto terapii kauzální. Zásady terapie se vztahují na všechny čtyři výše zmíněné běžné nemoci skladování.
Pacienti s Pompou jsou také léčeni infuzní terapií. Při tomto onemocnění je dodáván neexistující enzym „kyselá alfa-glukosidáza“, který pomáhá rozkládat glykogen, který se nahromadil v lysozomech svalů. U pacientů s onemocněním typu „mukopolysacharidóza typu I“ není lysosomální enzym „alfa-iduronidáza“ přítomen nebo není přítomen v dostatečném množství. Je to jedna z nejvzácnějších chorob skladování, ve kterých se molekuly cukru hromadí v orgánech a tkáních.
Pokud je proces normální, enzym štěpí mukopolysacharidy. Molekuly cukru mají dlouhý řetězec a podílejí se na vývoji podpůrné a pojivové tkáně, například kostí, kůže, kloubních tekutin a chrupavky. Pokud je normální nedostatek enzymu narušen v důsledku nedostatku enzymu, hromadí se v jednotlivých buňkách patologické glykosaminoglykany (GAG). Budoucí možnosti léčby jsou zaměřeny na užívání tablet.
Výhled a předpověď
Prognóza onemocnění při skladování je špatná. Bylo zjištěno, že genetická dispozice je příčinou zdravotní poruchy. Zákonné požadavky zakazují lékařům a vědcům měnit genetiku člověka. Z tohoto důvodu zůstává nemoc celoživotní a nemá žádnou šanci na uzdravení.
Ošetřující lékař se soustředí na léčbu symptomů, které se objevují. Pokud se neléčí, časem se budou zvyšovat různé stížnosti. Kostní systém je poškozen a vznikají problémy s orgány. V nejhorším případě dojde k selhání vnitřních orgánů a jejich funkce nakonec selže. To ohrožuje dotyčnou osobu předčasnou smrtí.
Výzva onemocnění spočívá v diagnostice. U velkého počtu pacientů se významné a silně vnímatelné příznaky objevují až později v životě. Výsledkem je, že genetická porucha zůstává dlouho bez povšimnutí a včasné léčení této choroby je obtížné. Čím je později stanovena diagnóza, tím je další průběh nepříznivější. V pokročilém stádiu onemocnění jsou již vnitřní orgány nebo klouby vážně poškozeny. Jsou nutné chirurgické zákroky a pokud nemoc nepříznivě progreduje, může život postižené osoby zachránit pouze jeden dárcovský orgán. Včasná léčba je proto nezbytná pro zlepšení prognózy.
prevence
Protože je to vrozená genetická vada, která brání expresi enzymu, nemůže být toto onemocnění léčeno preventivně. Poslední úspěchy v genetickém inženýrství však mohou poskytnout přístup v této oblasti.
Následná péče
S touto nemocí lidé trpí množstvím různých komplikací a onemocnění. Všichni mají zpravidla velmi negativní dopad na kvalitu života postižené osoby, takže diagnóza by měla být provedena velmi brzy.Čím dříve je konzultován lékař, tím lepší je další průběh tohoto onemocnění.
Závažnost tohoto onemocnění se může značně lišit, takže obecná předpověď není často možná. Dotčené osoby trpí vážným poškozením vnitřních orgánů. Ledviny a srdce jsou primárně postiženy, takže dítě může umřít v prvních několika dnech, pokud příznaky nejsou včas korigovány. Existují také usazeniny tuku v různých částech těla.
Obzvláště postižené jsou prsty a prsty na nohou, což může vést k významně snížené estetice postižené osoby. Ledviny a mozek jsou zpravidla poškozeny v dalším průběhu, takže dotyčná osoba v důsledku tohoto poškození zemře. Rodiče a příbuzní také často trpí depresí nebo jinými duševními poruchami v důsledku nemoci.
Můžete to udělat sami
Nemoci lysozomálního skladování často vyžadují intenzivní lékařskou péči. Často není dostatek příležitostí pro svépomoc. Rodiče postižených dětí často zažívají v domácím prostředí vážný stres, protože jejich dítě potřebuje stálou péči a pozornost.
Klinické obrázky jednotlivých chorob skladování se liší. Existují jednoduché i velmi obtížné formy. Jedním příkladem je Gaucherova choroba. Pomoc rodičů je často omezena na krmení těžce postiženého dítěte. V mírnějších případech může být délka života téměř normální. K odvrácení možných komplikací je však nutný neustálý lékařský dohled. Pravidelná fyzická aktivita je jednou z doprovodných terapií, které lze provádět i doma. Dále musí být provedeno důkladné vyšetření rakoviny. To vyžaduje neustálé návštěvy lékaře u jejich dítěte od rodičů. Totéž platí pro další choroby lysozomálního skladování.
V případě některých nemocí se kromě tělesného postižení mohou objevit i duševní poruchy, které stále vyžadují zvláštní podporu. U mírnějších forem některých nemocí, jako je Hunterova choroba, se zpočátku vyskytují pouze změny skeletu a dysmorfismus obličeje. Zde je však postižený pacient často schopen vést nezávislý život. Nicméně zde jsou také vyžadovány stálé lékařské prohlídky, aby se vyloučily možné komplikace, jako je srdeční selhání nebo respirační onemocnění. Pacient může psychickým stresem čelit psychickým stresům způsobeným psychologickým poradenstvím.
.jpg)





.jpg)



.jpg)