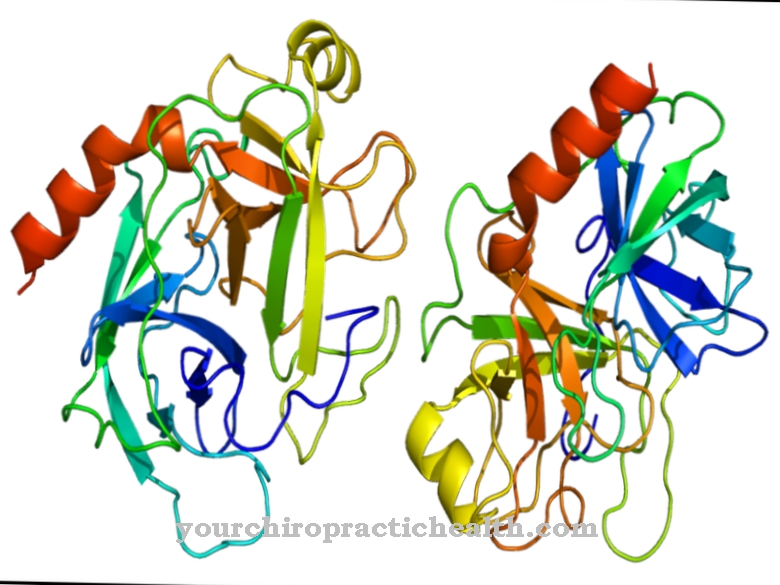
Lipoproteinová lipáza (LPL) patří do lipáz a hraje klíčovou roli v metabolismu lipidů. Je zodpovědný za rozdělení triglyceridů v chylomikronech a lipoproteinech s velmi nízkou hustotou (VLDL) na mastné kyseliny a monoacylglycerol. Uvolněné mastné kyseliny se používají ke generování energie nebo ke zvýšení tělesného tuku.
Co je lipoproteinová lipáza?
Lipoproteinová lipáza (LPL) je enzym, který je jednou z lipáz. Lipasy jsou zodpovědné za rozklad triglyceridů (triacylglyceroly) na mastné kyseliny a glycerin. Triglyceridy jsou estery trojného alkoholového glycerinu se třemi mastnými kyselinami, které se nazývají tuky nebo mastné oleje.
Tuky v potravě jsou absorbovány potravou a jsou nejprve štěpeny extracelulárními lipázami z pankreatu ve střevě. Některé triglyceridy se však dostávají do krevního řečiště absorpcí v tenkém střevě přes sérum, kde jsou vázány na lipoproteiny, které zaručují jejich schopnost transportu v krvi. Lipoproteinová lipáza je enzym, který štěpí triglyceridy navázané na lipoproteiny na mastné kyseliny a monoacylglycerol. Skládá se z 448 aminokyselin a je svou funkcí závislý na koenzymu apolipoproteinu C2.
Lipoproteinová lipáza je ve vodě rozpustný enzym, který se váže na endoteliální buňky krevních cév prostřednictvím určitých glykoproteinů (proteoglykany). Vyrábí se v játrech. Enzym katalyzuje hydrolýzu triglyceridů za vzniku dvou molekul mastných kyselin a jedné molekuly monoacylglycerolu. Apolipoproteiny jsou nosnými molekulami triglycerinů a umožňují jejich transport ve vodném prostředí. Apolipoprotein C2 také působí jako receptor lipoproteinové lipázy a aktivuje tak hydrolýzu triglyceridů.
Funkce, efekt a úkoly
Úkolem lipoproteinové lipázy je zcela katalyzovat rozklad tuků absorbovaných střevními buňkami v krvi. Nejprve jsou tuky v potravě rozloženy na mastné kyseliny a glycerin pomocí pankreatických lipáz v tenkém střevě. Další triglyceridy se dostávají do krve absorpcí přes tenké střevo a tam se vážou na lipoproteiny za vzniku komplexu lipid-protein.
To vytváří chylomikrony. Představují lipoproteinové částice o průměru 0,5 až 1 mikrometr, jejich hustota je menší než 1000 g / ml. Lipidové jádro obsahuje hlavně triglyceridy s malým množstvím esterů cholesterolu. Obal chylomikronů obsahující cholesterol obsahuje jako strukturní prvek fosfolipidy. Apolipoproteiny, na které jsou navázány triglyceridy, jsou nyní také uloženy v tomto obalu. Chylomikrony obsahují 90 procent triglyceridů. Dostávají se do krevního řečiště z tenkého střeva lymfatickým systémem. Triglyceridy se pomocí LPL rozkládají na mastné kyseliny a glycerin, zejména v kapilárách svalové a tukové tkáně.
Mastné kyseliny se používají buď ve svalové tkáni k výrobě energie, nebo v tukové tkáni k vytváření endogenních triglyceridů jako zásobního tuku. Po asi deseti hodinách potravinové abstinence již nelze v krvi detekovat žádné chylomikrony, protože triglyceridy se poté úplně rozloží. Dalšími složkami krve jsou tzv. VLDL (lipoprotein o velmi nízké hustotě). Tyto strukturní jednotky se uvolňují z jater a obsahují triglyceridy, fosfolipidy a cholesterol. VLDL transportuje tyto složky krevním oběhem z jater do jednotlivých orgánů.
Tímto způsobem jsou triglyceridy štěpeny lipoproteinovou lipázou a uvolňované mastné kyseliny jsou absorbovány tělními buňkami. Snížení triglyceridů převádí VLDL na LDL (lipoprotein s nízkou hustotou). LDL obsahují hlavně fosfolipidy, estery cholesterolu a lipoproteiny
Vzdělávání, výskyt, vlastnosti a optimální hodnoty
Lipoproteinová lipáza je syntetizována v játrech. Kromě pankreatických lipáz představuje další extracelulární lipázu, LPL se nachází na vnější straně membrán endoteliálních buněk různých orgánů, včetně tukových buněk. Tam je spojen s buněčnými membránami pomocí tzv. Proteoglykanů.
Je to však zvláště důležité pro endoteliální buňky krevních cév, protože zde může přímo řídit hydrolýzu triglyceridů v chylomikronech a VLDL. Heparin je injikován pro měření lipoproteázové aktivity. Heparin odstraňuje vazbu lipoproteinových lipáz z proteoglykanů, takže po injekci heparinu je zvýšená koncentrace volných lipoproteinových lipáz, která může být stanovena jejich aktivitou. Toto vyšetření může mimo jiné určit nedostatek lipoproteinové lipázy.
Nemoci a poruchy
Nedostatek lipoproteinové lipázy často vede k vážným zdravotním problémům. Pokud je lipoproteinová lipáza příliš nízká nebo pokud je její aktivita z důvodu genetické vady nedostatečná, lze triglyceridy v chylomikronech a VLDL rozložit pouze špatně nebo vůbec.
Deficit lipoproteinové lipázy může být primárně genetický, stejně jako sekundární k chemoterapii. Primární deficit LPL je vzácný a je způsoben autozomálně recesivní genetickou vadou. Vzniká takzvaná chylomikronémie, která je charakterizována mléčným krémovým sérem a je označována jako hyperlipidémie I. typu. Triglyceridy v chylomikronech se již nerozkládají. V důsledku toho se znovu a znovu objevují těžké pankreatidy s nesnášenlivostí mléka a bolestmi břicha.
Kromě toho se neustále vyvíjejí praskající xantomy a hepatomegalie. Jedinou možností léčby je nízkotučná strava a žádný alkohol. Toto onemocnění je často způsobeno mutacemi v genu LPL na chromozomu 8 nebo v genu APOC2. Sekundární forma hyperlipidémie typu I se obvykle vyskytuje s chemoterapií a má pouze dočasný charakter.



.jpg)



.jpg)






.jpg)
.jpg)

.jpg)