I buněčné onemocnění je lyozomální mukolipidóza. Příčinou onemocnění při skladování je mutace genu GNPTA s lokusem genu q23.3 na chromozomu 12. Symptomatické ošetření se provádí hlavně podáváním bisfosfonátů.
Co je to buněčná nemoc?

© red150770 - stock.adobe.com
Choroby skladování se vyznačují depozicí různých látek v buňkách a orgánech lidského těla. Je to heterogenní skupina nemocí, které lze rozdělit do několika podformulí. Kromě glykogenóz, mukopolysacharidóz a lipidóz rozlišuje medicína v závislosti na ukládané látce, sfingolipidózách, hemosiderózách a amyloidózách.
Nemoci lysozomálního skladování ovlivňují lysozomy. Jedná se o malé, membránou potažené buněčné organely v eukaryotech. Lysozomy jsou tvořeny Golgiho aparátem a jsou vybaveny hydrolytickými enzymy a fosfatázami. S pomocí svých enzymů by měli primárně trávit cizí látky a vlastní tělesné látky.
Onemocnění I-buněk je lyozomální mukolipidóza se dvěma různými podtypy. Leroy a DeMars tuto chorobu poprvé dokumentovali v 60. letech 20. století a poukazovali na její podobnost s mukopolysacharidózou typu I, známou jako Hurlerova choroba. Název nemoci pochází z fibroblastových inkluzí, tzv. Inkluzních buněk, v kůži pacienta.
příčiny
Příčinou onemocnění I-buněk je nedostatečná aktivita N-acetylglukosaminyl-1-fosfotransferázy. Omezená aktivita tohoto enzymu zabraňuje vstupu velké části lysozomálních enzymů do vnitřku lysozomu. Regulace lysozomálních enzymů je formována aktivitou fosfotransferázy.
Umožňuje syntézu třídicího signálu u zdravého organismu. Tento proces je u onemocnění I-buněk narušen. Proto neexistuje žádné značení manosou-6-fosfátem. Z tohoto důvodu již nejsou lysozomální enzymy řádně tříděny a migrují do extracelulární matrice nekontrolovaně přes plazmatickou membránu.
Důvodem je mutace v genu GNPTAB. Odstraňuje funkčnost N-acetylglukosaminyl-1-fosfotransferázy a tím schopnost katalyzovat syntézu manosy-6-fosfátu. Transport lysomálních enzymů je tak narušen. N-acetyl-glukosamin-1-fosfotransferáza se skládá z podjednotek alfa, beta a gama. Jsou kódovány na dvou genech.
Dědičné onemocnění I-buněk ovlivňuje gen GNPTA na chromozomu 12. V lokusu genu q23.3 je mutace. U vzácného onemocnění je uveden výskyt přibližně 0,3: 100 000. Dědičnost podléhá autosomální recesivní dědičnosti. Oba rodiče proto musí nést vadný gen, aby se nemoc mohla dále šířit.
Příznaky, onemocnění a příznaky
Ve většině případů lze příznaky onemocnění I-buněk pozorovat ihned po narození nebo nejpozději o několik měsíců později a jejich vlastnosti jsou podobné těm, které se vyskytují u Hurlerova syndromu. Na rozdíl od pacientů s Hurlerovým syndromem nevykazují pacienti s onemocněním I-buněk žádné vylučování mukopolysacharidů.
Jednotlivé příznaky nemoci podléhají velkému množství variací. Kornfeld a Sly shrnují klinické rysy kostry, vnitřních orgánů, očí, kůže, centrálního nervového systému a obličeje. Kostra je tak často ovlivněna kyphoskoliózou a dislokacemi kyčle.
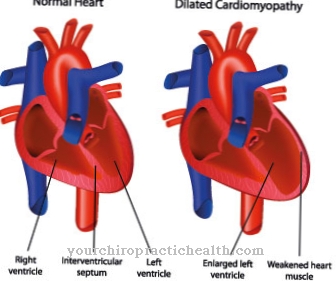
Rovněž mohou být přítomny klubové nohy, kloubní kontraktury a deformace obratlů. Totéž platí pro multiplex s krátkou postavou a dysostózou. Nemoc se může projevit ve vnitřních orgánech ve formě hepatosplenomegalie a kardioiomegalie nebo srdečních chorob. Tvář pacienta má hrubé rysy.
Typickými příznaky jsou exoftalmy, hyperplastické dásně nebo scaphocephaly. Charakteristické jsou také otevřená ústa a hluboce zapuštěný nos. Oči postižených mají často zákal rohovky nebo oteklé oční víčka. Kůže je silná a hrubá, s těžkou psychomotorikou nebo mentální retardací v centrálním nervovém systému.
Diagnóza a průběh nemoci
První podezření na diagnózu onemocnění I-buněk lze provést vizuální diagnostikou založenou na anamnéze. K potvrzení diagnózy lze použít biochemické stanovení aktivity lysozomálních enzymů v séru. Toto stanovení odhaluje absurdní vztah mezi intra- a extracelulární aktivitou.
Pro potvrzení diagnózy může být také stanovena aktivita fosfotransferázy ve fibroblastech. Vměstky odpovídají buď mukopolysacharidům, lipidům nebo oligosacharidům. Molekulární genetická diagnostika může rozptýlit zbývající pochybnosti. Pokud existuje vhodná anamnéza, lze onemocnění diagnostikovat také jako součást prenatální diagnostiky.
Vzhledem k nízké prevalenci je prenatální zpracování ve skutečnosti doporučeno pouze v případě rodinné dispozice. Průběh onemocnění závisí na symptomech v individuálním případě a není přímo předvídatelný. Většina pacientů však sotva přežije věk deseti. Mírnější formy rozvoje však nejsou v jednotlivých případech zcela vyloučeny.
Komplikace
Onemocnění I-buněk může vést k různým komplikacím a stížnostem. Jsou však rozpoznány pozdě, takže onemocnění I-buněk lze diagnostikovat pouze pozdě. Příznaky jsou relativně nekonzistentní, což často ztěžuje léčbu. To obvykle vede k nepohodlí a malformacím kůže, očí a vnitřních orgánů.
V nejhorším případě může zasažená osoba oslepnout nebo zemřít přímo na selhání orgánů. Kromě toho existuje výrazná krátká postava a také srdeční problémy. Oční víčka jsou často oteklá a je zde snížená inteligence a mentální retardace. Není neobvyklé, že postižená osoba bude kvůli retardaci závislá na pomoci jiných lidí v každodenním životě, aby se s ní vypořádala.
Kvalita života pacienta je značně snížena onemocněním I-buněk. Při léčbě nemoci zpravidla neexistují žádné zvláštní komplikace. Používají se léky a psychologické ošetření, které mohou zmírnit příznaky. Úplná a příčinná léčba tohoto onemocnění však není možná. Průměrná délka života je touto nemocí snížena.
Kdy byste měli jít k lékaři?
Onemocnění I buněk je obvykle diagnostikováno ihned po narození dítěte. Zda jsou nezbytná další léčebná opatření, závisí na typu a závažnosti příznaků. Menší malformace nemusí být nutně léčeny. Kluby a deformace obratlů jsou na druhé straně závažnými malformacemi, které je třeba léčit chirurgicky a léky. Rodiče by se měli okamžitě poradit s odborníkem, pokud to lékař v mateřské nemocnici tak již neučinil.
Pokud v důsledku reklamace dojde k nehodě nebo pádu, musí být dítě převezeno do nemocnice nebo rodiče by měli okamžitě zavolat pohotovostní službu. V případě závažných malformací, které mohou později ovlivnit psychiku dítěte v pozdějším věku, by měla být léčba doprovázena terapeutem. Onemocnění I-buněk proto vždy vyžaduje lékařské vyšetření. Správnou kontaktní osobou je pediatr nebo specialista na dědičné choroby. V případě zrakových poruch by měl být konzultován oftalmolog.
Lékaři a terapeuti ve vaší oblasti
Léčba a terapie
Onemocnění I-buněk je považováno za nevyléčitelné. Kauzální terapie proto neexistuje. Léčba je pouze symptomatická a podpůrná. Psychoterapeutická péče o postižené rodiny tvoří velkou část podpůrné terapie. Symptomatická terapie závisí na individuálním případě. Kostní příznaky se často léčí podáváním bisfosfonátů.
Tato léčiva jsou známá z léčby osteoporózy a mají vysokou afinitu k povrchu kosti. Zejména v oblasti resorpčních mezer se váží na kosti. Přitom inhibují osteoklasty degradující kost a tím snižují resorpci kosti. Léčiva jsou analogy pyrofosfátu s vazbou P-O-P obsahující uhlík.
Neprovádí se na nich enzymatická hydrolýza. Aminobisfosfonáty patří mezi nejnovější z těchto látek. Kromě toho jsou v Německu ze stejné skupiny léčiv schváleny alendronát, klodronát, etidronát, ibandronát, pamidronát a risendronát. Totéž platí pro tiludronát a zoledronát.
Kromě těchto léčiv mohou být transplantáty kostní dřeně také použity k léčbě onemocnění I-buněk. Úspěch této léčby byl omezen pouze v předchozích případech. Genové terapie jsou nyní zkoumány jako nový terapeutický přístup pro genové defekty. Genové terapie ukázaly počáteční úspěch ve zvířecích modelech. Doposud se nemohly v praxi použít na lidech. Tento vztah se však v budoucnu pravděpodobně změní.
Zde najdete své léky
➔ Léky proti bolestiVýhled a předpověď
Onemocnění I-buněk je dědičné onemocnění, které dosud nebylo léčeno symptomaticky. Prognóza je tedy negativní. Ačkoli symptomy mohou být významně sníženy časnou terapií, onemocnění I-buněk téměř vždy trvá vážný průběh.
Krátká postava a poškození vnitřních orgánů a hlavy již značně snižují délku života. Kromě toho mohou malformace obličeje, kůže a očí snížit očekávanou délku života, ale především také kvalitu života postižené osoby. Někteří z postižených dosáhnou věku 40 nebo 50 let, ale většina z nich zemře v dětství nebo dospívání.
Pokud se onemocnění I buněk neléčí, nemocní často umírají v prvních několika letech života. Prognóza je tedy spíše negativní. Vyhlídka na život bez relativně příznaků je nicméně dána, pokud je pacient léčen jako součást komplexní terapie a je-li to nutné, je umístěno v ústavu pro tělesně postižené osoby. Fyzioterapie a terapeutická opatření mohou z dlouhodobého hlediska významně zlepšit pohodu pacienta.
prevence
I-buněčnému onemocnění lze před plánováním rodiny zabránit molekulárně genetickým testem. V rámci prenatální diagnostiky se mohou nastávající rodiče také rozhodnout ukončit těhotenství.
Následná péče
Ve většině případů nemají osoby postižené onemocněním I-buněk k dispozici žádná nebo jen velmi malá následná opatření. Toto onemocnění musí být lékařem rozpoznáno co nejdříve, aby bylo možné zabránit dalšímu zhoršení příznaků. Protože se jedná o geneticky podmíněnou chorobu, mělo by být vždy provedeno genetické vyšetření a konzultace, pokud chcete mít děti, aby se zabránilo přenosu nemoci I-buněk na potomky.
Většina pacientů je na toto onemocnění závislá na různých lécích. Je důležité zajistit správné dávkování a pravidelné užívání léků. Pokud je něco nejasné, existují vedlejší účinky nebo máte-li jakékoli otázky, měli byste se nejprve poradit s lékařem.
Podobně mnoho nemocných potřebuje psychologickou podporu s touto nemocí, i když milující diskuse s rodiči nebo příbuznými mohou mít pozitivní vliv na průběh nemoci. Dotčená osoba potřebuje pomoc a podporu v každodenním životě od své vlastní rodiny. V mnoha případech onemocnění I-buněk významně omezuje nebo snižuje střední délku života postižené osoby.
Můžete to udělat sami
Pacienti trpící onemocněním I-buněk se mohou uchýlit k různým konzervativním a alternativním léčebným metodám. Konzervativní terapie se zaměřuje na zmírnění příznaků a onemocnění.
Použití pomocných látek, jako jsou berle nebo ortopedické vložky, může zpomalit průběh příslušných malformací a tím také zmírnit bolest. Léky pomáhají zmírňovat bolest a mohou být doplněny alternativními opatřeními, jako je masáž nebo akupunktura. Alternativní terapie by měly být předem konzultovány s odpovědným lékařem. Lékař může být schopen obrátit pacienta přímo na homeopat nebo může poskytnout další tipy, jak léčit příslušný symptom.
Protože onemocnění I-buněk je obvykle fatální, navzdory všem léčebným možnostem, je třeba vyhledat terapeutickou radu. Jejich obavy by se neměly týkat pouze postižených. Příbuzní a přátelé obvykle také potřebují podporu při řešení nemoci a jejího možného negativního výsledku. Účast na svépomocné skupině je také možností pro pacienta a jeho příbuzné. Kontakt s jinými nemocnými pomáhá s přijetím nemoci a často mohou i jiní pacienti navrhnout další léčebná opatření a strategie pro každodenní život s I-buněčnou chorobou.
.jpg)










.jpg)